岩田 觉
东京大学 研究生院情报理工学系研究科 教授
北海道大学 化学反应创成研究基地(WPI-ICReDD)特任教授
ERATO副研究总括(2019年度~2024年度)
大城 泰平
北海道大学 化学反应创成研究基地(WPI-ICReDD)特任副教授
松冈 和
北海道大学 研究生院理学研究院 助教
原渕 祐
北海道大学 化学反应创成研究基地(WPI-ICReDD)特任教授
前田 理
北海道大学 研究生院理学研究院 教授
化学反应创成研究基地(WPI-ICReDD)主任
2019年起担任ERATO研究总括
化学已从反复实验的试错学科,逐步进化至在计算机上的模拟,近年来,融合量子化学计算和信息科学等,预测和控制化学反应的研究趋势正在加速。北海道大学研究生院理学研究院的前田理教授和东京大学研究生院信息理工学系研究科的岩田觉教授等人,正在跨学科尝试通过捕捉化学反应中原子的运动全貌,不断设计出有用的未知化学反应,由此创造出新的“智能”。
从试错学科迈向新领域
求解方程式,解析物质微观结构与性质
人类与化学的关系,与人类文明紧密相连。人类自古就懂得如何让身边物质的产生变化,并通过系统性地积累知识,推动化学发展至今。然而,化学是一门以实验为基础的学科,要制作出有用的材料和药品,需要反复进行大量实验,耗费诸多的时间和成本。
由北海道大学前田理教授率领的ERATO“前田化学反应创成智能项目”,旨在将试错学科的化学,提升为项目名称所示的“化学反应创成智能”这一新学术领域。为了预测和控制化学反应,需从理论上阐明反应是如何发生的。其中备受关注的方法是量子化学计算。量子化学计算通过近似求解在原子世界支配事物的量子力学基本方程——薛定谔方程(图1),来解析原子和分子的结构、性质以及反应性。
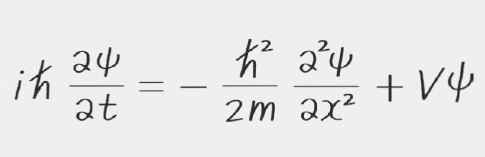
图1 1926年奥地利物理学家埃尔温·薛定谔发表的量子力学基本方程。
薛定谔方程发表于1926年,但在当时,求解它来计算实际的化学反应非常困难。近年来,随着求解方法和计算机的进步,对现实化学反应的模拟已成为可能。如果能够通过计算机的计算来预测过去必须通过试验尝试才能知晓的化学实验结果,会在加速材料开发、削减实验成本和废弃物等方面获得巨大优势。
前田在硕士课程期间接触到了量子化学计算,被其难度所吸引,并因此开启了他作为研究者的职业生涯。前田回顾当时说道:“指导教授发现了我喜欢编程和思考算法的天赋,便引导我进入了量子化学计算领域。自己也是一经尝试便喜欢上了它。”
超越人类认知的自动探索反应路径
凭借“虚拟力”引发变化
既然能用量子化学计算预测原子和分子的结构、性质及反应性,似乎任何材料和药品都能立刻制造出来,但实际上并非如此简单。这是因为,预测化学反应的主角——原子运动的全貌极其困难。
前田项目所追求的“化学反应创成智能”,通过融合量子化学计算、信息科学、材料信息学(Materials Informatics)等技术,来预测化学反应中原子运动的全貌。其基础是前田等人领先世界开发出来的人工力诱导反应(Artificial Force Induced Reaction, AFIR)法。
要预测未知运动的全貌,就需要一种能够超越人类所能思考极限的自动探索化学反应行进路径的计算方法。AFIR法在分子间或分子内部分结构之间施加虚拟的人工力,引发结构变化。通过系统性地重复该操作,计算将给定的反应物转化为未知生成物的路径。通过解析如此得到的反应路径网络,即可预测未知反应(图2)。

图2 AFIR法概念图(左)与AFIR法获得的反应路径网络示例(右)。通过计算将反应物A和B转化为未知生成物X的路径,并解析得到的网络,就有可能预测反应。
出处:Satoshi M.et al., Springer, Cham,2020
在化学反应中,分子与分子存在易于反应的方向和位置,当改变方向的同时让分子相互靠近时,分子会自行排列到易于反应的方向和位置上。前田解释AFIR法的关键时说道:“通过巧妙地控制施加推力的方式,就能够有效地找到反应结构。”
通过“逆合成解析”预测最佳原料
无偿公开路径数据库
前田项目的具体目标是利用AFIR法,针对各种反应物和催化剂的组合计算反应路径网络,并创建数据库。此外,还将致力于构建一个基于数据库的、快速设计适合合成目标化学物质的化学反应系统。在实现上述目标的过程中,目前已取得了多项突破性成果。
首先在2022年,确立了完全不使用有机化学知识以及实验数据、仅基于量子化学计算的“逆合成解析”方法。并对某种天然有机化合物使用逆合成解析方法,证明了能够逆合成出已经被实验证实的起始原料(图3)。这一成果是向量子化学计算的终极目标之一——“从零开始预测新的化学反应,并通过实验实现”迈出的一步,这是通过结合AFIR法和速率常数矩阵缩约(RCMC)法实现的。
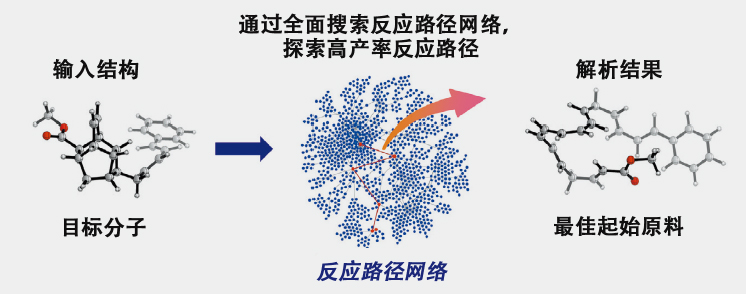
图3 利用涵盖所有可能反应路径的反应路径网络,进行逆向追溯化学反应路径的“逆合成解析”示意图,可高精度预测最佳起始原料。
AFIR法虽能进行全面探索,但反应路径会有多处分支,形成复杂且庞大的网络,其解析需要庞大的计算量。对此,RCMC法是一种通过关注的时间尺度,逐步缩约无需区分的部分区域,从而高效计算化学反应的方法。将其与AFIR法结合,在反应路径的分支中,仅通过RCMC法筛选实际可能发生的分支进行探索,修剪掉不必要的分支,能够减少计算量。
接下来在2023年,收录有AFIR法创建的化学反应路径数据的数据库平台“SCAN(Searching Chemical Actions and Networks)”作为开源资源进行了无偿公开(图4)。实现了仅通过点击即可执行数据检索、可视化和数据解析,即使不是信息科学专家也能解析复杂的化学反应路径数据。
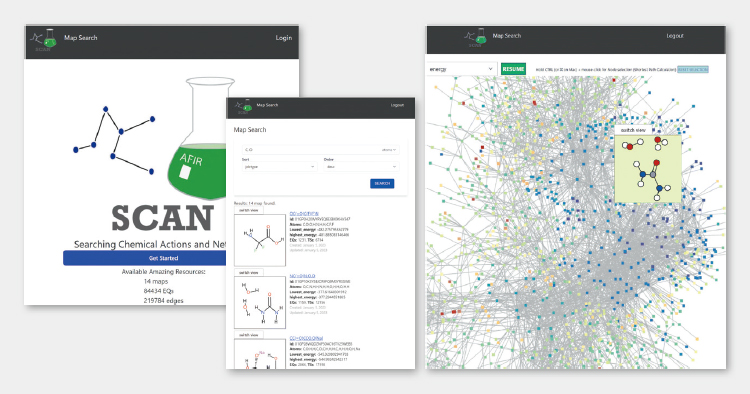
图4 收录AFIR法生成的化学反应路径数据的数据库平台“SCAN”。网页(https://scan.sci.hokudai.ac.jp/)已公开,仅通过点击即可执行化学反应路径的检索、可视化、探索和设计等所有操作。
确立过渡金属催化剂开发指针
引入微分与自动最优化方法
最后,2024年发表在《美国化学会志》的成果是该项目的集大成之一。该成果确立了过渡金属催化反应中催化剂的新设计指针,证明了以往需要根据大量实验数据估算的设计指针,仅通过量子化学计算和微分计算即可求得。
过渡金属催化剂由过渡金属和与其结合的被称为“配体”的有机分子组成,其催化性能因种类而异。因此,要想在温和条件下实现高效的化学反应,寻找性能最佳的配体至关重要。然而,配体数量庞大,若采用既往的实验或计算方法逐一尝试的话将耗费过多时间。
于是,前田项目中“量子化学团队”的松冈和助教与“最优化团队”的大城泰平特任副教授,着眼于配体对催化性能的电子配置效应和空间位阻效应,构建了一种将这些性质作为少数输入参数进行计算的机制。松冈表示:“我们将配体赋予催化剂的特性视为连续参数,通过微分求得参数形成的‘斜率’,再运用连续优化,就能比以往方法更显著高效地进行最优化。”
在松冈和大城开发的“虚拟配体辅助优化法(Virtual Ligand-Assisted Optimization, VLAO法)”中,量子化学计算使用的是将实际配体模型化后的虚拟配体(图5)。通过微分估算这些模型化配体的电子和空间性质对催化性能的影响,可以自动最优化配体的性质。由此便能够获得高效推进目标反应的线索。
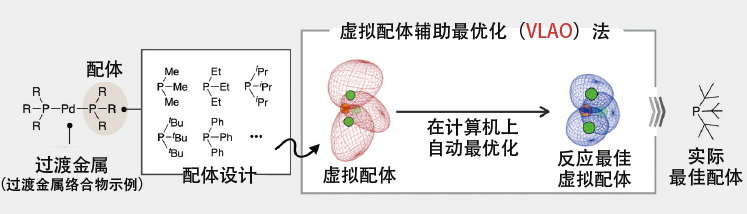
图5 在以配体结合的过渡金属为催化剂的过渡金属催化反应量子化学计算中,利用模型化实际配体的虚拟配体,可通过微分计算估算性质对反应性的影响并进行最优化。
松冈等人还在实际使用钯催化剂的反应中,成功在无需任何实验的情况下找到了优于现有配体的新配体。期待此次在计算机上实现预测的方法,能够解决以往在开发时间和成本方面的各种问题,为社会做出巨大贡献。
集结数学等不同领域的专家
解决仅靠化学难以解决的问题
前田项目的独特之处在于,集结了化学、数学、信息科学、材料学等不同专业领域的成员,通过发挥各自的专长,挑战仅靠化学专家无法解决的难题。除了前面介绍的松冈所属的“量子化学团队”和大城所属的“最优化团队”外,还有构建数据库的“化学信息团队”、根据AFIR法发现的反应路径实际进行化学合成实验的“有机合成团队”、利用机器人实现高速实验的“机器人合成团队”等共7个团队协同合作。
其中,以前田为中心的化学领域和以东京大学岩田觉教授为中心的数学领域是项目的支柱。岩田担任本项目的副研究总括,通过离散优化等数学方法,与大城共同为化学反应路径网络的解析和RCMC法的高速化做出较大的贡献。
尽管化学和数学同属理科领域,但其内容完全不同,使用的专业术语也不同。因此,大城和松冈表示,在项目启动初期,彼此的沟通都很困难。数学研究学者大城谈到合作的成果时说:“起初连单词都不懂,但在反复对话中,我了解到化学领域存在许多在数学上很有趣的问题。数学难以解决的问题在化学中却得到了解决,理解对方的背景让人很有收获。”
另一方面,化学研究学者松冈回顾道:“化学中有很多经验法则,但在与数学专家交流的过程中,我领受到了严格定义的数学世界的有趣之处。同时学到了如果能提取出关键部分并将其转化为数学问题,会带来巨大的好处。”前田列举了项目贡献者、担任研究总括助理的北海道大学原渕祐特任教授。他称赞道:“起初成员间沟通困难,是原渕先生耐心地将大家连接起来。RCMC法最初也是由原渕先生和大城先生深入讨论,运用数学的方法才获得了近期的成果。”
可以说,该项目是跨领域合作取得成效的典范。岩田认为今后融合性研究、跨学科研究会越来越多,他期待道:“虽然很辛苦,但正因如此才取得了巨大成果。尤其是连接化学和数学的人才还非常少,希望参与项目的研究学者今后能取得更多成果。”(TEXT:石井英男,PHOTO:岛本绘梨佳)

原文:JSTnews 2025年6月号
翻译:JST客观日本编辑部